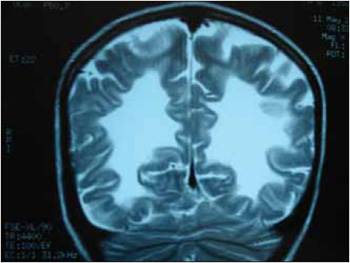
A adrenoleucodistrofia (ADL) é uma doença genética com padrão de herança ligado ao X , que consiste numa alteração do metabolismo dos peroxissomos, ocasionando um acúmulo de ácidos graxos de cadeia muito longa (AGCML) constituídos de 24 e 26 átomos de carbono no organismo sobretudo no cérebro e nas glândulas adrenais. Tal acúmulo está associado à desmielinização dos axônios afetando a transmissão dos impulsos nervosos e a insuficiência adrenal. Afeta quase exclusivamente o sexo masculino com início dos sintomas entre 4 e 10 anos e incidência estimada de 1:25000 homens. A manifestação clínica da doença consiste inicialmente em alterações de comportamento, da audição, da visão, da fala, da escrita, memória, da marcha, distúrbios adrenais e nos casos mais avançados cursa com hipertonia generalizada, perda das funções cognitivas, motoras, convulsões e disfagia. O diagnóstico é confirmado dosando-se os níveis plasmáticos dos AGCML, Ressonância Magnética mostrando lesões desmielinizantes com distribuição em "asa de mariposa" na substância branca parieto-occipital bi-hemisférica, eletromiografia compatível com polineuropatia tipo mielinopático, pesquisa laboratorial para insuficiência adrenal e cariótipo cujo gene defeituoso é o ABCD1 localizado no lócus X9-28 do cromossomo X (2, 3, 4,5,6).


DISCUSSÃO
A ADL é uma doença genética rara incluída no grupo das leucodistrofias e que afeta o cromossomo X, sendo uma herança ligada ao sexo de caráter recessivo transmitida por mulheres portadoras e que afeta fundamentalmente ho-mens. O gene defeituoso é responsável pela codificação de uma enzima denominada ligase acil CoA gordurosa, que é encontrada na membrana dos peroxissomos e está relacionada ao transporte de ácidos graxos para o interior dessa estrutura celular. O gene defeituoso ocasiona uma mutação nessa enzima, os AGCML ficam impedidos de entrar nos peroxissomos e se acumulam no interior celular. Os mecanismos precisos através dos quais os AGCML ocasionam a destruição na bainha de mielina ainda são desconhecidos. As possibilidades de descendência a partir de uma mulher portadora da ALD são 25% de chance de nascer um filho normal; 25% de nascer um filho afetado; 25% de nascer uma filha normal; 25% de nascer uma filha portadora heterozigota. As chances de descendência para um homem afetado, se tiver filhas, serão todas portadoras da doença; e se tiver filhos, serão todos normais; (1). A clínica da doença é muito variável mas a manifestação inicial mais frequente é a mudança de comportamento da criança apresentando isolamento social e défict de atenção. Habitualmente nestes casos é questionada a audição do paciente que pode variar de normal a graus variados de perda neurossensorial melhor verificados com BERA já que o paciente pode apresentar algum prejuízo cognitivo prejudicando a realização da audiometria convencional. O BERA quando alterado mostrauma diminuição da amplitude e no tempo de condução das ondas sugerindo uma patologia retrococlear e alterações diversas de limiar.
No nosso paciente em especial, o BERA mostrou-se normal, com audiometria tonal de difícil realização que evidenciou perda auditiva neurossensorial moderada. Nesse instante poderia se tratar de uma perda auditiva central ou uma doença neurológica ou psicossomática como autismo, por exemplo. Provavelmente os potenciais auditivos de média e longa latência, como o P300, poderiam nos auxiliar na detecção de alterações a nível de córtex cerebral.
Outros sinais como disartria, dismetria , bradilalia, disgrafia ou alterações na marcha apontarão para a hipótese de doença neurodegenerativa a qual deve ser investigada e diagnosticada. Na ADL o diagnóstico é auxiliado pela Ressonância Magnética que mostra lesões desmielinizantes com distribuição em asa de mariposa na substância branca parieto-occipital bi-hemisférica, dosagem dos níveis plasmáticos dos AGCML aumentados, eletromiografia compatível com polineuropatia tipo mielinopático, pesquisa laboratorial para insuficiência adrenal e cariótipo. Após o diagnóstico é importante uma equipe multiprofissional para acompanhamento e reabilitação desses doentes já que se trata de uma doença evolutiva. Em fases mais avançadas cursam com graus variados de disfagia neurogênica diagnoticadas através de videoendoscopia da deglutição ou videodeglutograma. A terapia nestes estágios consiste em manobras de deglutição ou gastrostomia se alimentação oral não é segura ou inadequada.
Não existe uma terapia definida para a ADL até o momento, segundo estudos a dieta livre de AGCML como espinafre, queijo e carne vermelha, associada ao azeite ou "óleo de Lorenzo" têm obtido êxito especialmente se administrada no início ou antes da aparição dos sintomas. O tratamento da disfunção adrenal, através da administração de hormônios e atualmente os transplantes de medula são modalidades de tratamento adotadas na ADL com grau de sucesso na evolução da doença muito variável na literatura. Muitas vezes, o grande intervalo de tempo entre o início dos sintomas e o diagnóstico, prejudica o tratamento e aponta para um pequeno conhecimento dos médicos sobre o assunto (7,8,9,10). O otorrinolaringologista, em especial, deve conhecer esta doença pois é, habitualmente, um dos primeiros profissionais a serem consultados podendo então, contribuir para um diagnóstico precoce ao encaminhá-los ao neuropediatra e ao geneticista beneficiando assim, não só o paciente mas também os familiares na identificação dos portadores e no aconselhamento genético.
COMENTÁRIOS FINAIS
A ADL é uma doença rara, neurodegenerativa, que necessita de diagnóstico precoce para um melhor prognóstico. Não existe uma terapia definitiva porem o tratamento com Óleo de Lorenzo tem obtido êxito associado a um acompanhamento multiprofissional. O otorrinolaringologista habitualmente é um dos primeiros profissionais a entrar em contato com esses pacientes, devendo encaminhá-los precocemente para diagnóstico e acompanhá-los até fases mais avançadas onde a disfagia se faz presente.
Henrique é portador da X-Ald,usou o Óleo de Lorenzo,fêz a dieta restrita em ácidos graxos e 01 ano depois o transplante de medula.
ResponderExcluirDescobrimos em 1998.Médicos Osmário Jorge Salles e Fernando Kok.Hoje Henrique esta curado.Criei um blog adrenoleucodistrofiax-ald.blogspot.com para contar o caminho que percorremos para conquistar a cura de Henrique